Trajectory inference for hematopoiesis in mouse¶
Reconstructing myeloid and erythroid differentiation for data of Paul et al. (2015).
[1]:
import numpy as np
import pandas as pd
import matplotlib.pyplot as pl
from matplotlib import rcParams
import scanpy as sc
[2]:
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
results_file = './write/paul15.h5ad'
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3)) # low dpi (dots per inch) yields small inline figures
scanpy==1.4.6.dev1+g1404638 anndata==0.7rc2.dev9+g5928e64 umap==0.3.8 numpy==1.16.3 scipy==1.3.0 pandas==0.25.3 scikit-learn==0.22 statsmodels==0.10.0 python-igraph==0.7.1 louvain==0.6.1
[3]:
adata = sc.datasets.paul15()
WARNING: In Scanpy 0.*, this returned logarithmized data. Now it returns non-logarithmized data.
[4]:
adata
[4]:
AnnData object with n_obs × n_vars = 2730 × 3451
obs: 'paul15_clusters'
uns: 'iroot'
Let us work with a higher precision than the default ‘float32’ to ensure exactly the same results across different computational platforms.
[5]:
adata.X = adata.X.astype('float64') # this is not required and results will be comparable without it
Preprocessing and Visualization¶
Apply a simple preprocessing recipe.
[6]:
sc.pp.recipe_zheng17(adata)
running recipe zheng17
normalizing counts per cell
finished (0:00:00)
extracting highly variable genes
finished (0:00:00)
normalizing counts per cell
finished (0:00:00)
finished (0:00:00)
[7]:
sc.tl.pca(adata, svd_solver='arpack')
computing PCA with n_comps = 50
finished (0:00:00)
[8]:
sc.pp.neighbors(adata, n_neighbors=4, n_pcs=20)
sc.tl.draw_graph(adata)
computing neighbors
using 'X_pca' with n_pcs = 20
finished: added to `.uns['neighbors']`
'distances', distances for each pair of neighbors
'connectivities', weighted adjacency matrix (0:00:03)
drawing single-cell graph using layout 'fa'
finished: added
'X_draw_graph_fa', graph_drawing coordinates (adata.obsm) (0:00:10)
[9]:
sc.pl.draw_graph(adata, color='paul15_clusters', legend_loc='on data')
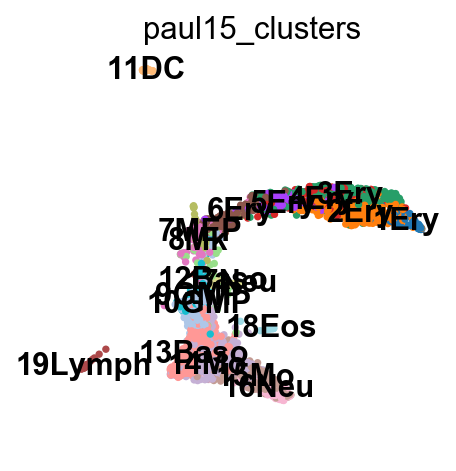
This looks like a mess.
Optional: Denoising the graph¶
To denoise the graph, we represent it in diffusion map space (and not in PCA space). Computing distances within a few diffusion components amounts to denoising the graph - we just take a few of the first spectral components. It’s very similar to denoising a data matrix using PCA. The approach has been used in a couple of papers, see e.g. Schiebinger et al. (2017) or Tabaka et al. (2018). It’s also related to the principles behind MAGIC Dijk et al. (2018).
Note
This is not a necessary step, neither for PAGA, nor clustering, nor pseudotime estimation. You might just as well go ahead with a non-denoised graph. In many situations (also here), this will give you very decent results.
[10]:
sc.tl.diffmap(adata)
sc.pp.neighbors(adata, n_neighbors=10, use_rep='X_diffmap')
computing Diffusion Maps using n_comps=15(=n_dcs)
computing transitions
finished (0:00:00)
eigenvalues of transition matrix
[1. 1. 0.9989277 0.99671 0.99430376 0.98939794
0.9883687 0.98731077 0.98398703 0.983007 0.9790806 0.9762548
0.9744365 0.9729161 0.9652972 ]
finished: added
'X_diffmap', diffmap coordinates (adata.obsm)
'diffmap_evals', eigenvalues of transition matrix (adata.uns) (0:00:00)
computing neighbors
finished: added to `.uns['neighbors']`
'distances', distances for each pair of neighbors
'connectivities', weighted adjacency matrix (0:00:00)
[11]:
sc.tl.draw_graph(adata)
drawing single-cell graph using layout 'fa'
finished: added
'X_draw_graph_fa', graph_drawing coordinates (adata.obsm) (0:00:10)
[12]:
sc.pl.draw_graph(adata, color='paul15_clusters', legend_loc='on data')
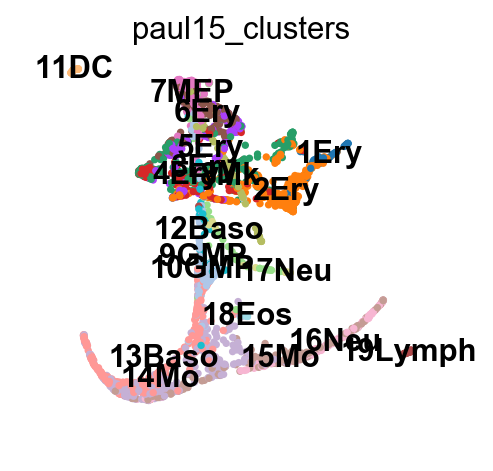
This still looks messy, but in a different way: a lot of the branches are overplotted.
Clustering and PAGA¶
Note that today, we’d use sc.tl.leiden - here, we use sc.tl.louvain the sake of reproducing the paper results.
[13]:
sc.tl.louvain(adata, resolution=1.0)
running Louvain clustering
using the "louvain" package of Traag (2017)
finished: found 25 clusters and added
'louvain', the cluster labels (adata.obs, categorical) (0:00:00)
Annotate the clusters using marker genes.
| cell type | marker |
|---|---|
| HSCs | Procr |
| Erythroids | Gata1, Klf1, Epor, Gypa, Hba-a2, Hba-a1, Spi1 |
| Neutrophils | Elane, Cebpe, Ctsg, Mpo, Gfi1 |
| Monocytes | Irf8, Csf1r, Ctsg, Mpo |
| Megakaryocytes | Itga2b (encodes protein CD41), Pbx1, Sdpr, Vwf |
| Basophils | Mcpt8, Prss34 |
| B cells | Cd19, Vpreb2, Cd79a |
| Mast cells | Cma1, Gzmb, CD117/C-Kit |
| Mast cells & Basophils | Ms4a2, Fcer1a, Cpa3, CD203c (human) |
For simple, coarse-grained visualization, compute the PAGA graph, a coarse-grained and simplified (abstracted) graph. Non-significant edges in the coarse- grained graph are thresholded away.
[14]:
sc.tl.paga(adata, groups='louvain')
running PAGA
finished: added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns) (0:00:00)
[15]:
sc.pl.paga(adata, color=['louvain', 'Hba-a2', 'Elane', 'Irf8'])
--> added 'pos', the PAGA positions (adata.uns['paga'])
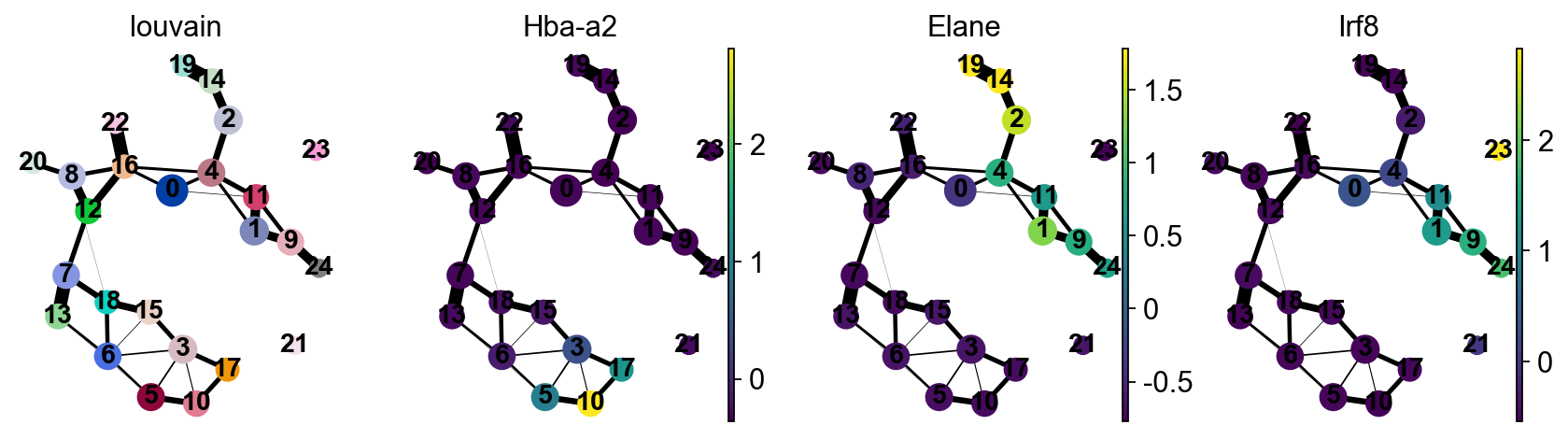
[16]:
sc.pl.paga(adata, color=['louvain', 'Itga2b', 'Prss34', 'Cma1'])
--> added 'pos', the PAGA positions (adata.uns['paga'])
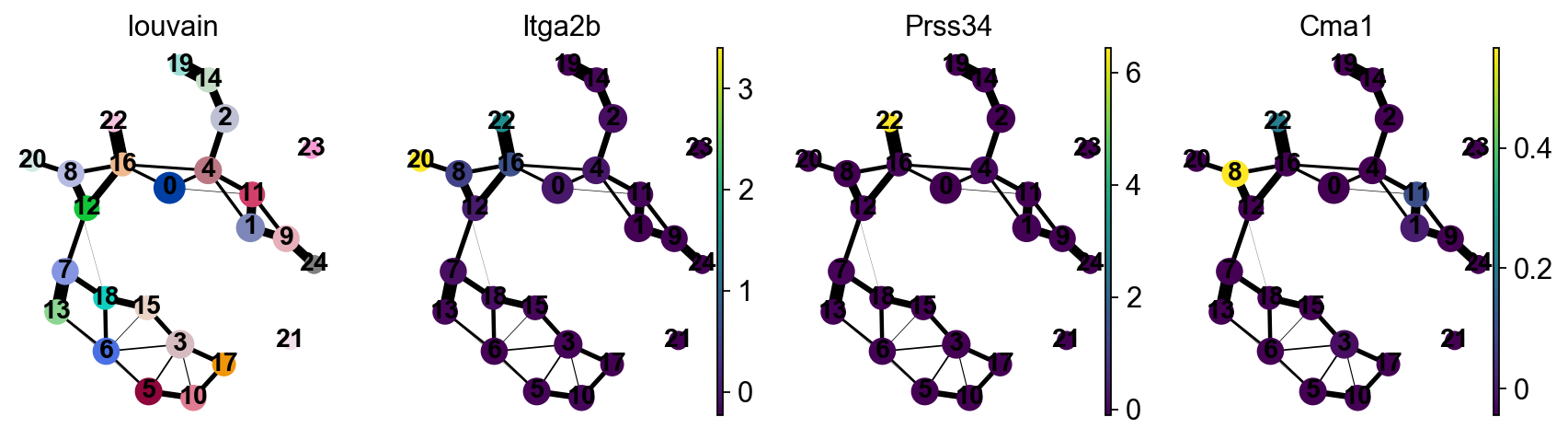
Actually annotate the clusters — note that Cma1 is a Mast cell marker and only appears in a small fraction of the cells in the progenitor/stem cell cluster 8, see the single-cell resolved plot below.
[17]:
adata.obs['louvain'].cat.categories
[17]:
Index(['0', '1', '2', '3', '4', '5', '6', '7', '8', '9', '10', '11', '12',
'13', '14', '15', '16', '17', '18', '19', '20', '21', '22', '23', '24'],
dtype='object')
[18]:
adata.obs['louvain_anno'] = adata.obs['louvain']
[19]:
adata.obs['louvain_anno'].cat.categories = ['0', '1', '2', '3', '4', '5', '6', '7', '8', '9', '10/Ery', '11', '12',
'13', '14', '15', '16/Stem', '17', '18', '19/Neu', '20/Mk', '21', '22/Baso', '23', '24/Mo']
Let’s use the annotated clusters for PAGA.
[20]:
sc.tl.paga(adata, groups='louvain_anno')
running PAGA
finished: added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns) (0:00:00)
[21]:
sc.pl.paga(adata, threshold=0.03, show=False)
--> added 'pos', the PAGA positions (adata.uns['paga'])
[21]:
<matplotlib.axes._axes.Axes at 0x13ea01ef0>

Recomputing the embedding using PAGA-initialization¶
The following is just as well possible for a UMAP.
[22]:
sc.tl.draw_graph(adata, init_pos='paga')
drawing single-cell graph using layout 'fa'
finished: added
'X_draw_graph_fa', graph_drawing coordinates (adata.obsm) (0:00:10)
Now we can see all marker genes also at single-cell resolution in a meaningful layout.
[23]:
sc.pl.draw_graph(adata, color=['louvain_anno', 'Itga2b', 'Prss34', 'Cma1'], legend_loc='on data')
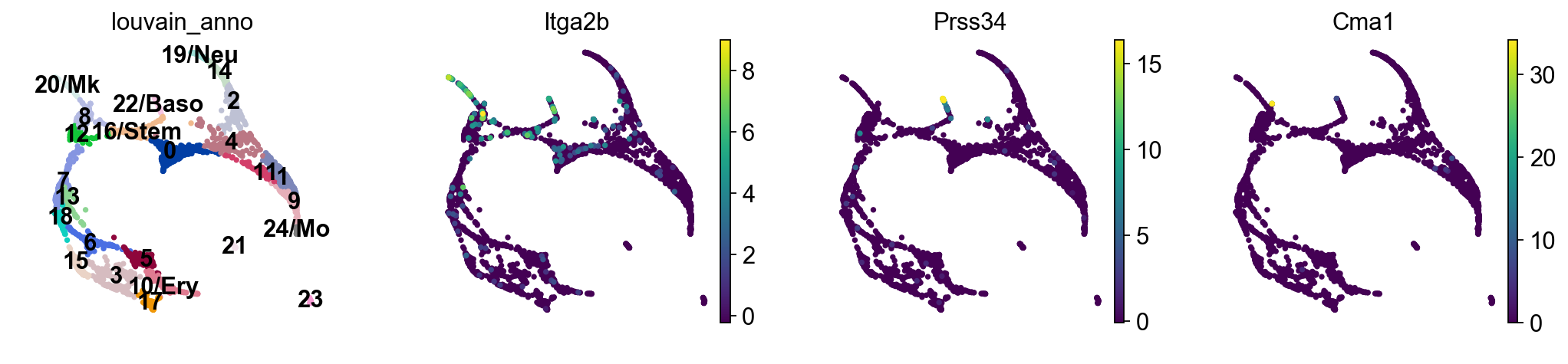
Choose the colors of the clusters a bit more consistently.
[24]:
pl.figure(figsize=(8, 2))
for i in range(28):
pl.scatter(i, 1, c=sc.pl.palettes.zeileis_28[i], s=200)
pl.show()
[25]:
zeileis_colors = np.array(sc.pl.palettes.zeileis_28)
new_colors = np.array(adata.uns['louvain_anno_colors'])
[26]:
new_colors[[16]] = zeileis_colors[[12]] # Stem colors / green
new_colors[[10, 17, 5, 3, 15, 6, 18, 13, 7, 12]] = zeileis_colors[[5, 5, 5, 5, 11, 11, 10, 9, 21, 21]] # Ery colors / red
new_colors[[20, 8]] = zeileis_colors[[17, 16]] # Mk early Ery colors / yellow
new_colors[[4, 0]] = zeileis_colors[[2, 8]] # lymph progenitors / grey
new_colors[[22]] = zeileis_colors[[18]] # Baso / turquoise
new_colors[[19, 14, 2]] = zeileis_colors[[6, 6, 6]] # Neu / light blue
new_colors[[24, 9, 1, 11]] = zeileis_colors[[0, 0, 0, 0]] # Mo / dark blue
new_colors[[21, 23]] = zeileis_colors[[25, 25]] # outliers / grey
[27]:
adata.uns['louvain_anno_colors'] = new_colors
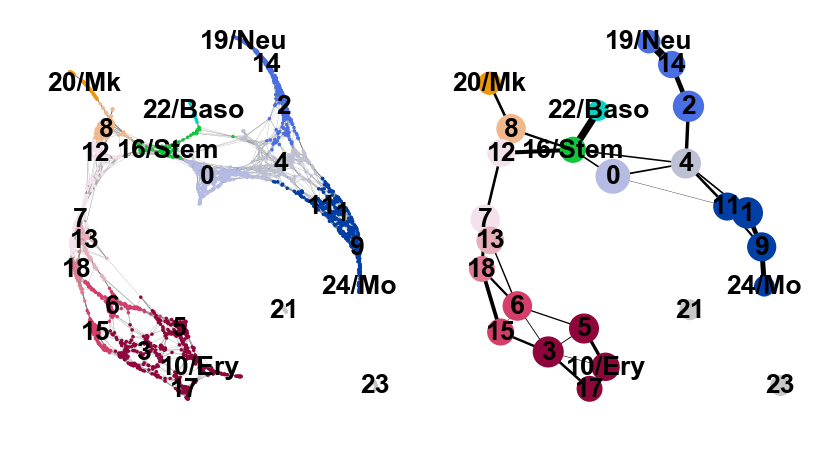
And add some white space to some cluster names. The layout shown here differs from the one in the paper, which can be found here. These differences, however, are only cosmetic. We had to change the layout as we moved from a randomized PCA and float32 to float64 precision.
[28]:
sc.pl.paga_compare(
adata, threshold=0.03, title='', right_margin=0.2, size=10, edge_width_scale=0.5,
legend_fontsize=12, fontsize=12, frameon=False, edges=True, save=True)
--> added 'pos', the PAGA positions (adata.uns['paga'])
WARNING: saving figure to file figures/paga_compare.pdf
Reconstructing gene changes along PAGA paths for a given set of genes¶
Choose a root cell for diffusion pseudotime.
[29]:
adata.uns['iroot'] = np.flatnonzero(adata.obs['louvain_anno'] == '16/Stem')[0]
[30]:
sc.tl.dpt(adata)
computing Diffusion Pseudotime using n_dcs=10
finished: added
'dpt_pseudotime', the pseudotime (adata.obs) (0:00:00)
Select some of the marker gene names.
[31]:
gene_names = ['Gata2', 'Gata1', 'Klf1', 'Epor', 'Hba-a2', # erythroid
'Elane', 'Cebpe', 'Gfi1', # neutrophil
'Irf8', 'Csf1r', 'Ctsg'] # monocyte
Use the full raw data for visualization.
[32]:
adata_raw = sc.datasets.paul15()
sc.pp.log1p(adata_raw)
sc.pp.scale(adata_raw)
adata.raw = adata_raw
WARNING: In Scanpy 0.*, this returned logarithmized data. Now it returns non-logarithmized data.
[33]:
sc.pl.draw_graph(adata, color=['louvain_anno', 'dpt_pseudotime'], legend_loc='on data')
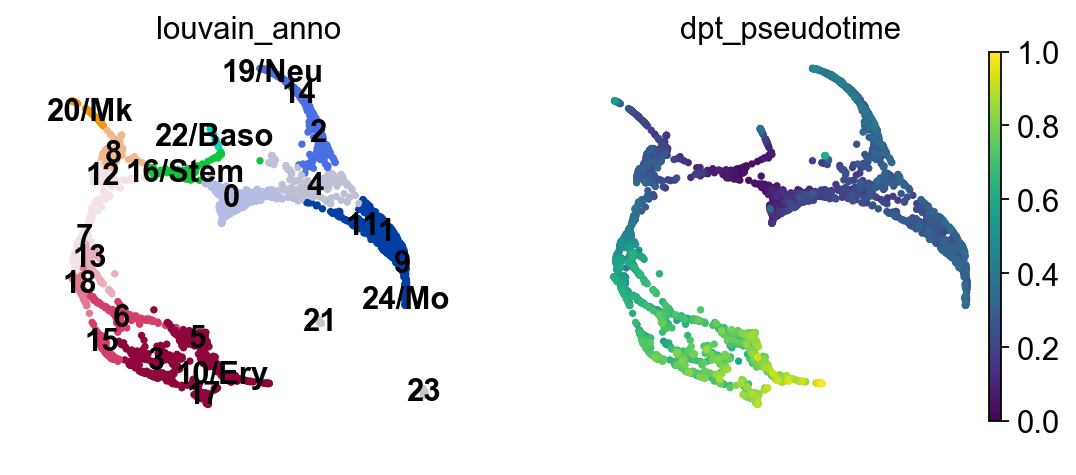
[34]:
paths = [('erythrocytes', [16, 12, 7, 13, 18, 6, 5, 10]),
('neutrophils', [16, 0, 4, 2, 14, 19]),
('monocytes', [16, 0, 4, 11, 1, 9, 24])]
[35]:
adata.obs['distance'] = adata.obs['dpt_pseudotime']
[36]:
adata.obs['clusters'] = adata.obs['louvain_anno'] # just a cosmetic change
[37]:
adata.uns['clusters_colors'] = adata.uns['louvain_anno_colors']
[38]:
!mkdir write
mkdir: write: File exists
[39]:
_, axs = pl.subplots(ncols=3, figsize=(6, 2.5), gridspec_kw={'wspace': 0.05, 'left': 0.12})
pl.subplots_adjust(left=0.05, right=0.98, top=0.82, bottom=0.2)
for ipath, (descr, path) in enumerate(paths):
_, data = sc.pl.paga_path(
adata, path, gene_names,
show_node_names=False,
ax=axs[ipath],
ytick_fontsize=12,
left_margin=0.15,
n_avg=50,
annotations=['distance'],
show_yticks=True if ipath==0 else False,
show_colorbar=False,
color_map='Greys',
groups_key='clusters',
color_maps_annotations={'distance': 'viridis'},
title='{} path'.format(descr),
return_data=True,
show=False)
data.to_csv('./write/paga_path_{}.csv'.format(descr))
pl.savefig('./figures/paga_path_paul15.pdf')
pl.show()
