Danger
Please access this document in its canonical location as the currently accessed page may not be rendered correctly: Plotting with Marsilea
Plotting with Marsilea#
Marsilea is a visualization library that allows user to create composable visualization in a declarative way.
You can use it to create many scanpy plots with easy customization.
Let’s first load the PBMC datdaset
from __future__ import annotations
import numpy as np
import scanpy as sc
pbmc = sc.datasets.pbmc3k_processed().raw.to_adata()
pbmc
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'percent_mito', 'n_counts', 'louvain'
var: 'n_cells'
uns: 'draw_graph', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_tsne', 'X_umap', 'X_draw_graph_fr'
obsp: 'distances', 'connectivities'
Define the cells and markers that we want to draw
cell_markers = {
"CD4 T cells": ["IL7R"],
"CD14+ Monocytes": ["CD14", "LYZ"],
"B cells": ["MS4A1"],
"CD8 T cells": ["CD8A"],
"NK cells": ["GNLY", "NKG7"],
"FCGR3A+ Monocytes": ["FCGR3A", "MS4A7"],
"Dendritic cells": ["FCER1A", "CST3"],
"Megakaryocytes": ["PPBP"],
}
cells, markers = [], []
for c, ms in cell_markers.items():
cells += [c] * len(ms)
markers += ms
uni_cells = list(cell_markers.keys())
cell_colors = [
"#568564",
"#DC6B19",
"#F72464",
"#005585",
"#9876DE",
"#405559",
"#58DADA",
"#F85959",
]
cmapper = dict(zip(uni_cells, cell_colors, strict=True))
Import Marsilea
import marsilea as ma
import marsilea.plotter as mp
Heatmap#
Here is the minimum example to create a heatmap with Marsilea, it does nothing besides create a heatmap. You can adjust the main plot size by setting height and width, the unit is inches.
We will start adding components to this main plot step by step.
exp = pbmc[:, markers].X.toarray()
m = ma.Heatmap(exp, cmap="viridis", height=3.5, width=3)
m.render()
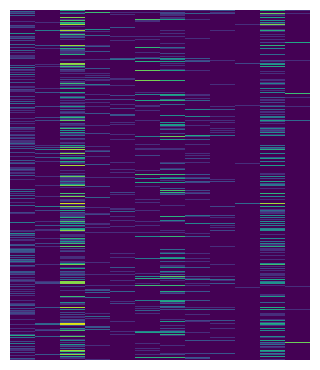
To replicate the scanpy heatmap, we can first divide the heatmap by cell types.
The group_rows method can group heatmap by group labels, the first argument is used to label the row, the order defines the display order of
each cell type from top to bottom.
m.group_rows(pbmc.obs["louvain"], order=uni_cells)
m.render()
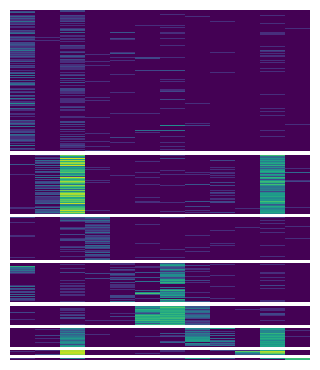
Now we can label each chunks with cell types and the columns with marker names.
You can use add_left or add_* to add a plotter to anyside of your main plot. In Marsilea, a plot instance is called plotter.
When you add a plotter, you can easily adjust its size and the padding between adjcent plot using size and pad.
# Create plotters
chunk = mp.Chunk(uni_cells, rotation=0, align="center")
colors = mp.Colors(list(pbmc.obs["louvain"]), palette=cmapper)
label_markers = mp.Labels(markers)
# Add to the heatmap
m.add_left(colors, size=0.1, pad=0.1)
m.add_left(chunk)
m.add_top(label_markers, pad=0.1)
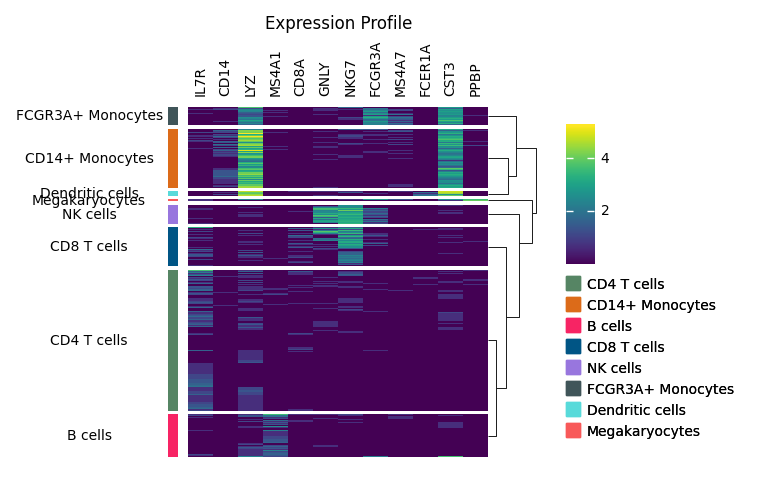
m.render()
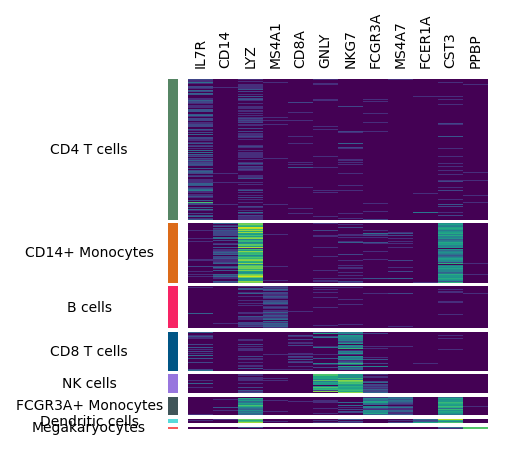
You may want to add dendrogram to display the similarity among cell types.
You can use add_dendrogram, we will add it to the right side, but you can also add it to the left side if you want.
m.add_dendrogram("right", add_base=False)
m.render()
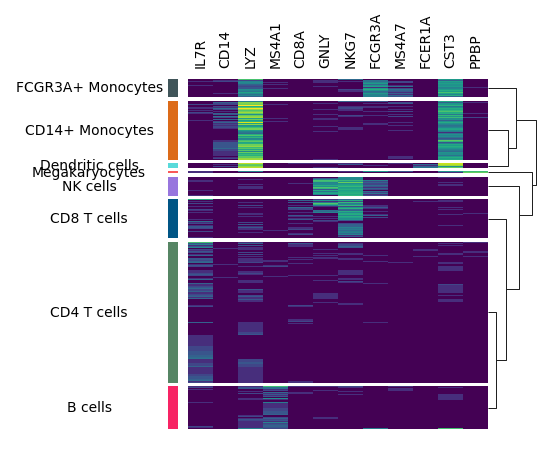
The legend is still mising, you can use add_legends to add all legends at once. Marsilea will automatically layout all the legends.
In the end, you can you add_title to add a title.
m.add_legends()
m.add_title("Expression Profile")
m.render()
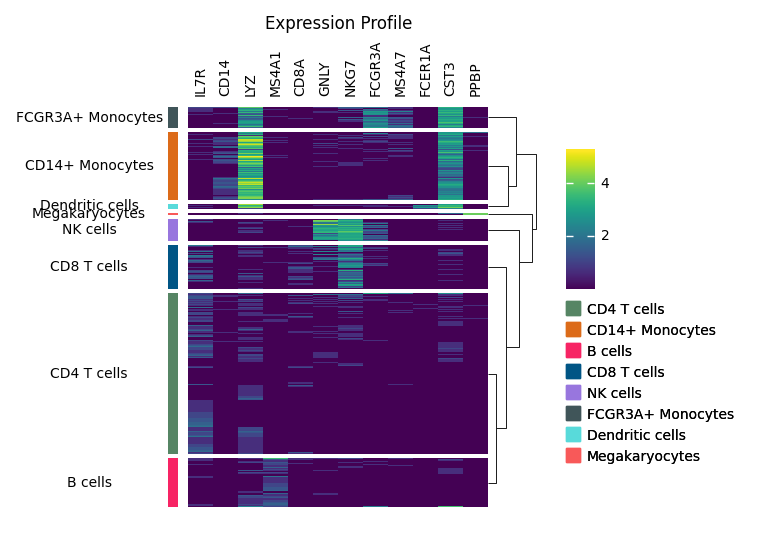
OK, let’s wrap up all the code in below. There are few things you should notice in Marsilea:
Always call
render()in the end to actually render the plot.The order of
add_*operation decides the order of plotters. Butgroup_rowsandgroup_colscan be called anytime.
m = ma.Heatmap(exp, cmap="viridis", height=4, width=3)
m.group_rows(pbmc.obs["louvain"], order=uni_cells)
m.add_left(
mp.Colors(list(pbmc.obs["louvain"]), palette=cmapper),
size=0.1,
pad=0.1,
)
m.add_left(mp.Chunk(uni_cells, rotation=0, align="center"))
m.add_top(mp.Labels(markers), pad=0.1)
m.add_dendrogram("right", add_base=False)
m.add_legends()
m.add_title("Expression Profile")
m.render()
Now that we’ve covered some basics of Marsilea, we’ll see how it can be used to create custom plots similar to scanpy’s existing methods:
agg = sc.get.aggregate(pbmc[:, markers], by="louvain", func=["mean", "count_nonzero"])
agg.obs["cell_counts"] = pbmc.obs["louvain"].value_counts()
agg
AnnData object with n_obs × n_vars = 8 × 12
obs: 'louvain', 'cell_counts'
var: 'n_cells'
layers: 'mean', 'count_nonzero'
agg_exp = agg.layers["mean"]
agg_count = agg.layers["count_nonzero"]
agg_cell_counts = agg.obs["cell_counts"].to_numpy()
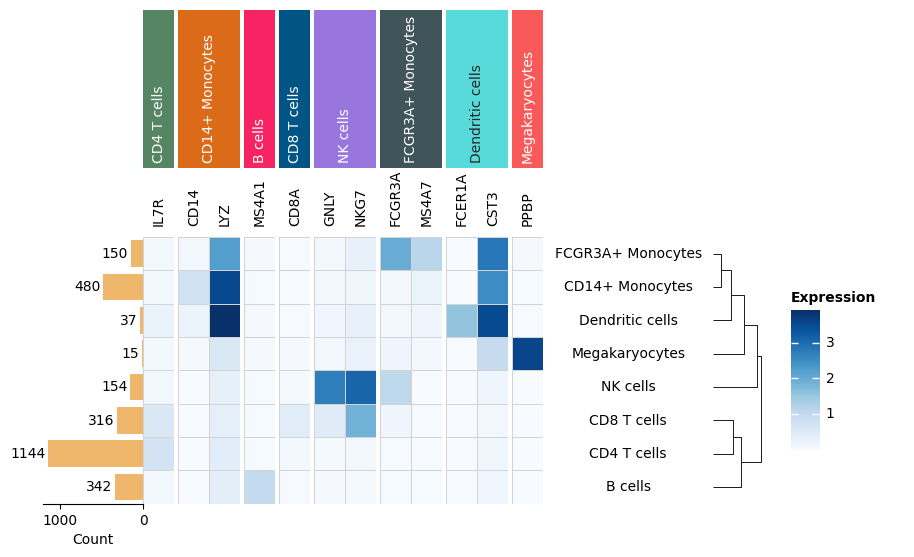
Matrixplot#
h, w = agg_exp.shape
m = ma.Heatmap(
agg_exp,
height=h / 3,
width=w / 3,
cmap="Blues",
linewidth=0.5,
linecolor="lightgray",
label="Expression",
)
m.add_right(mp.Labels(agg.obs["louvain"], align="center"), pad=0.1)
m.add_top(mp.Labels(markers), pad=0.1)
m.group_cols(cells, order=uni_cells)
m.add_top(mp.Chunk(uni_cells, fill_colors=cell_colors, rotation=90))
m.add_left(mp.Numbers(agg_cell_counts, color="#EEB76B", label="Count"))
m.add_dendrogram("right", pad=0.1)
m.add_legends()
m.render()
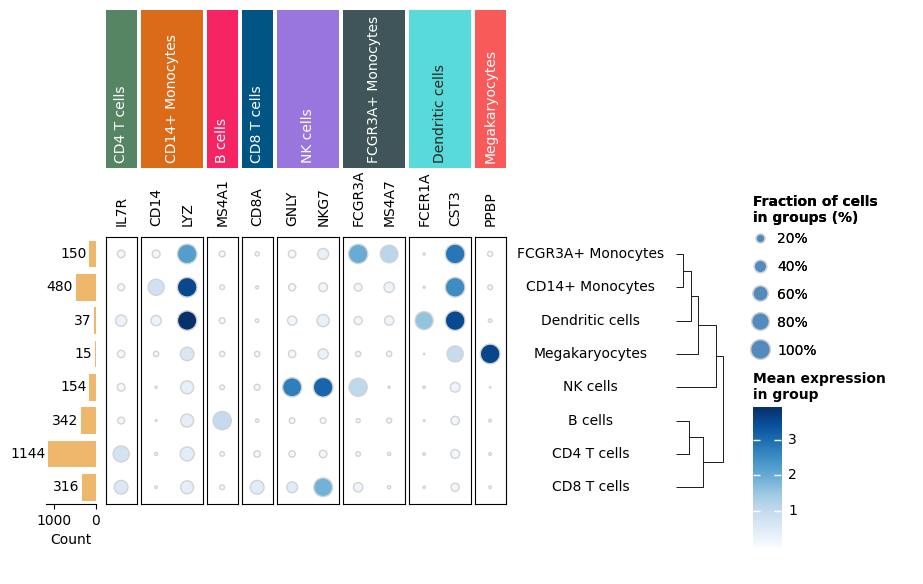
Dot plot#
size = agg_count / agg_cell_counts[:, np.newaxis]
m = ma.SizedHeatmap(
size=size,
color=agg_exp,
cluster_data=size,
height=h / 3,
width=w / 3,
edgecolor="lightgray",
cmap="Blues",
size_legend_kws=dict(
colors="#538bbf",
title="Fraction of cells\nin groups (%)",
labels=["20%", "40%", "60%", "80%", "100%"],
show_at=[0.2, 0.4, 0.6, 0.8, 1.0],
),
color_legend_kws=dict(title="Mean expression\nin group"),
)
m.add_top(mp.Labels(markers), pad=0.1)
m.add_top(mp.Chunk(uni_cells, fill_colors=cell_colors, rotation=90))
m.group_cols(cells, order=uni_cells)
m.add_right(mp.Labels(agg.obs["louvain"], align="center"), pad=0.1)
m.add_left(mp.Numbers(agg_cell_counts, color="#EEB76B", label="Count"), size=0.5, pad=0.1)
m.add_dendrogram("right", pad=0.1)
m.add_legends()
m.render()
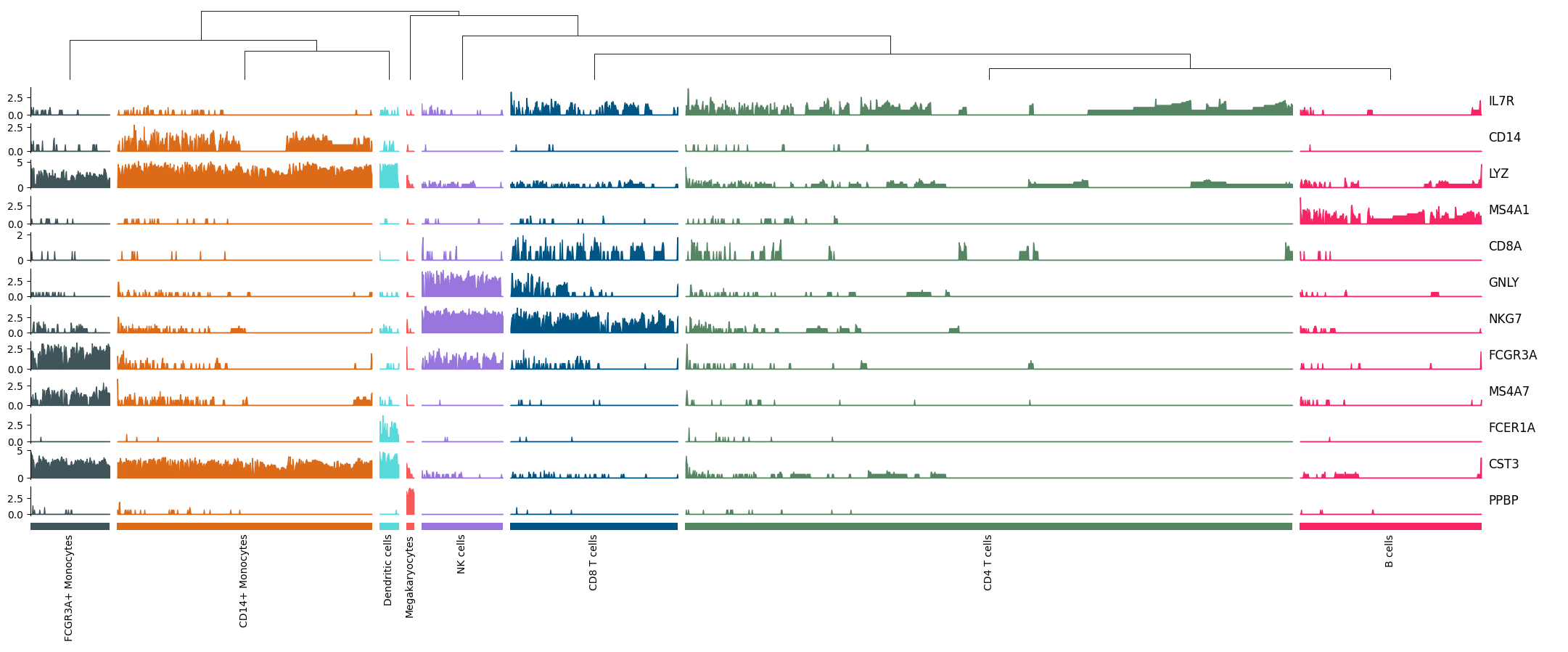
Tracksplot#
tp = ma.ZeroHeightCluster(exp.T, width=20)
tp_anno = ma.ZeroHeight(width=1)
tp.group_cols(pbmc.obs["louvain"], order=uni_cells, spacing=0.005)
tp.add_dendrogram("top", add_base=False, size=1)
for row in exp.T:
area = mp.Area(
row,
add_outline=False,
alpha=1,
group_kws={"color": cell_colors},
)
tp.add_bottom(area, size=0.4, pad=0.1)
tp.add_bottom(mp.Colors(pbmc.obs["louvain"], palette=cmapper), size=0.1, pad=0.1)
tp.add_bottom(mp.Chunk(uni_cells, rotation=90))
(tp + 0.1 + tp_anno).render()
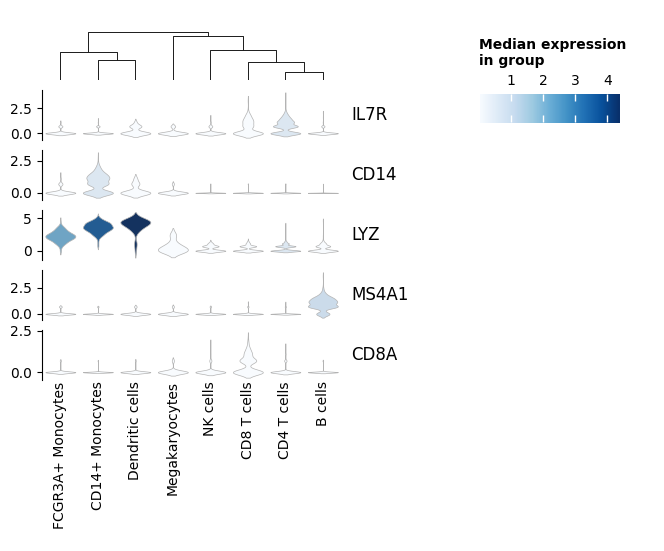
Stacked Violin#
import pandas as pd
from matplotlib.cm import ScalarMappable
from matplotlib.colors import Normalize
gene_data = []
cdata = []
for row, gene_name in zip(exp.T, markers[:5], strict=True):
# Transform data to wide-format, marsilea only supports wide-format
pdata = (
pd.DataFrame({"exp": row, "cell_type": pbmc.obs["louvain"]}) # noqa: PD010
.reset_index(drop=True)
.pivot(columns="cell_type", values="exp")
)
gene_data.append([gene_name, pdata])
cdata.append(pdata.median())
# Create color mappable
cdata = pd.concat(cdata, axis=1).T
vmin, vmax = np.asarray(cdata).min(), np.asarray(cdata).max()
sm = ScalarMappable(norm=Normalize(vmin=vmin, vmax=vmax), cmap="Blues")
sv = ma.ZeroHeightCluster(agg_exp.T, width=3)
sv_anno = ma.ZeroHeight(width=1)
for gene_name, pdata in gene_data:
# Calculate the color to display
palette = sm.to_rgba(pdata.median()).tolist()
sv.add_bottom(
mp.Violin(
pdata,
inner=None,
linecolor=".7",
linewidth=0.5,
density_norm="width",
palette=palette,
),
size=0.5,
pad=0.1,
legend=False,
)
sv_anno.add_bottom(mp.Title(gene_name, align="left"), size=0.5, pad=0.1)
sv.add_bottom(mp.Labels(cdata.columns))
sv.add_dendrogram("top")
# To fake a legend
sv.add_bottom(
mp.ColorMesh(
cdata,
cmap="Blues",
cbar_kws={"title": "Median expression\nin group", "orientation": "horizontal"},
),
size=0,
)
comp = sv + 0.1 + sv_anno
comp.add_legends()
comp.render()
More information#
Other plots are possible with Marsilea by creating new plotter. See Marsilea’s documention to learn how.
import session_info
session_info.show(dependencies=True)
Click to view session information
----- anndata 0.10.6 marsilea 0.3.5 matplotlib 3.8.3 numpy 1.26.4 pandas 2.2.0 scanpy 1.10.1 session_info 1.0.0 -----
Click to view modules imported as dependencies
PIL 10.2.0 anyio NA appnope 0.1.3 arrow 1.3.0 asttokens NA attr 23.2.0 attrs 23.2.0 babel 2.14.0 certifi 2024.02.02 cffi 1.16.0 charset_normalizer 3.3.2 colorama 0.4.6 comm 0.2.1 cycler 0.12.1 cython_runtime NA dateutil 2.8.2 debugpy 1.8.0 decorator 5.1.1 defusedxml 0.7.1 exceptiongroup 1.2.0 executing 2.0.1 fastjsonschema NA fqdn NA h5py 3.10.0 idna 3.6 ipykernel 6.29.0 isoduration NA jedi 0.19.1 jinja2 3.1.3 joblib 1.3.2 json5 NA jsonpointer 2.4 jsonschema 4.21.1 jsonschema_specifications NA jupyter_events 0.9.0 jupyter_server 2.12.5 jupyterlab_server 2.25.2 kiwisolver 1.4.5 legacy_api_wrap NA legendkit 0.3.4 llvmlite 0.42.0 markupsafe 2.1.5 matplotlib_inline 0.1.6 mpl_toolkits NA natsort 8.4.0 nbformat 5.9.2 numba 0.59.1 overrides NA packaging 23.2 parso 0.8.3 patsy 0.5.6 pexpect 4.9.0 platformdirs 4.1.0 prometheus_client NA prompt_toolkit 3.0.43 psutil 5.9.8 ptyprocess 0.7.0 pure_eval 0.2.2 pyarrow 15.0.0 pydev_ipython NA pydevconsole NA pydevd 2.9.5 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.17.2 pyparsing 3.1.1 pythonjsonlogger NA pytz 2023.3.post1 referencing NA requests 2.31.0 rfc3339_validator 0.1.4 rfc3986_validator 0.1.1 rpds NA scipy 1.12.0 seaborn 0.13.2 send2trash NA six 1.16.0 sklearn 1.4.0 sniffio 1.3.0 sphinxcontrib NA stack_data 0.6.3 statsmodels 0.14.1 threadpoolctl 3.2.0 tornado 6.4 traitlets 5.14.1 typing_extensions NA uri_template NA urllib3 2.1.0 wcwidth 0.2.13 webcolors 1.13 websocket 1.7.0 yaml 6.0.1 zmq 25.1.2
----- IPython 8.20.0 jupyter_client 8.6.0 jupyter_core 5.7.1 jupyterlab 4.0.11 ----- Python 3.10.0 | packaged by conda-forge | (default, Nov 20 2021, 02:27:15) [Clang 11.1.0 ] macOS-14.2.1-arm64-arm-64bit ----- Session information updated at 2024-04-19 15:23